In September 2023 the US Food and Drug Administration (FDA) issued industry guidance on cybersecurity for medical devices to better protect patients, hospitals, and the broader healthcare system from cyber-attacks. Medical device and in vitro diagnostic (IVD) manufacturers will be required to implement significant changes to Design Control and Quality Management practices and procedures to comply with the new regulations. The FDA guidance is a result of years of study and a well-documented increase in malicious attacks on hospitals and other healthcare distribution centers. Indeed, ransomware attacks are now commonplace among hospital systems (large and small) with heavy financial and even patient care consequences. Healthcare distribution systems are unique in their vulnerability to cyber-attacks due to a history of prioritizing patient care and patient outcomes over cybersecurity threats and a reliance on a vast array of tools and devices to manage care.
The Intersection of Cybersecurity and Medical Devices
The past fifteen years have seen remarkable increases in software-enabled smart medical devices and a shift towards an Internet of Things (IoT) healthcare distribution architecture. These trends are responses to strong market demand for smart devices’ benefits, including wider patient access, more effective use of data, better patient experiences, and better patient outcomes. However, these benefits carry increased risks of malicious attacks on healthcare organizations by criminals who exploit vulnerable devices to target individual patient medical records, disrupt operations, ransom data, or enter networks through backdoors to move freely throughout an enterprise. Manufacturers of medical devices must do their part to remedy a situation that is increasing in frequency and severity by improving the quality of their products.
FDA’s Cybersecurity Guidance for Medical Device Manufacturers
The impacts of the FDA’s cybersecurity guidance are only now being appreciated. The scope of the regulations is broad and includes all device software that stores, transfers or analyzes data. Therefore, any medical or diagnostic device with upgradeable software, a USB port, or even compact disc technology is now considered a connected device and is subject to updated regulations. It is important to understand that as of September 2023 any company, whether a startup or a Fortune 500 medical device or IVD manufacturer, developing devices and technology for FDA-regulated markets must update their product development procedures to address the new standards. This means medical device and IVD developers must now add resources and effort to quality management and design controls. It is also significant that the new guidance encompasses not only modern wirelessly connected and IoT technology but also the huge toolbox of existing products already in hospitals. The impacts of this reality could be even more substantial. They will play out as the industry faces balancing legacy technology’s security risks and upgrading systems’ costs.
The Guidance reflects FDA’s recommendations for information to be included in premarket submissions for Basic and Enhanced Documentation Levels. This recommended information should demonstrate that planning, requirements, risk assessment, design reviews, traceability, change management, testing plans and results, and other aspects of good software engineering for device software functions were employed, to support a conclusion that the device software function was appropriately designed, verified, and validated
FDA, Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions
What Changed at Triple Ring?
In preparation for meeting the design requirements implied by the new medical device cybersecurity rules, Triple Ring’s Quality, Systems Engineering, and Software Engineering teams have completed training on the guidance, have updated quality management processes, and have begun implementing device designs and documentation that will support successful FDA submissions for our clients. The new practices augment a robust and comprehensive quality management system by adding a threat modeling methodology called STRIDE. We have implemented the STRIDE methodology to systematically assess vulnerabilities and mitigate threats throughout the device design lifecycle. We follow a structured process, outlined below, for all our client projects requiring FDA submissions.
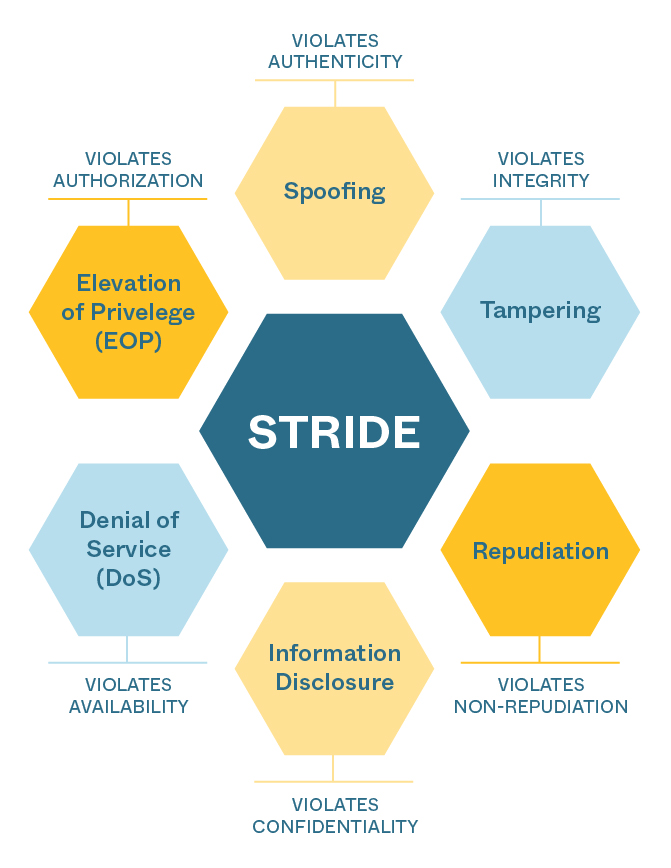
Figure: STRIDE framework for assessing, mitigating, and designing devices resistant to cybersecurity attacks.
STRIDE Process
- Identify and document device design elements requiring protection: The use of data flow diagrams (DFD) in parallel with the product architecture allows Triple Ring to decompose any device design to help the FDA understand key elements within the design, core functionality, external connections and how information flows through the device. From software, firmware, and embedded code to wireless communication, and data transfer hardware, the subsystems and components that require threat protection are cataloged in data flow tables.
- Analyze each element using STRIDE methodology: At the subsystem and component level, we analyze the device design for potential vulnerabilities using STRIDE.
- Document potential vulnerabilities and threats: We fully characterize device designs for each STRIDE category and document our findings in detail. By defining the ecosystem and system DFDs, the product can be used to identify assets that threat actors may utilize to probe at vulnerabilities of the product. Triple Ring is well-versed in defining asset and data flow tables in combination with security use cases to analyze the ecosystem and interactions with the device to identify threat events that may occur from element to element.
- Rank the likelihood and impact of threats, and develop mitigations: We then go through a process of identifying threats in accordance with the AAMI TIR-57 Security Risk Process. We quantify the likelihood and severity of each threat using FMEA tools (e.g. decision matrices), prioritize threats according to likelihood and severity, and then identify and develop mitigation strategies (e.g. encryption, access controls, or input validation).
- Test, verify, and iterate: Triple Ring uses the generalized System Science approach to engineering development, which prescribes an iterative process for analyzing, specifying, designing, building, and testing. Gaps identified in our threat modeling analyses are used as inputs for further iterations of the device design and mitigation-specific vulnerabilities.
- Include threat assessment in all FDA submissions: We then include all findings in regulatory documentation and ultimately in the device Design History File (DHF). FDA now requires cybersecurity threat assessments in most medical device and IVD submissions. Clear identification, understanding, characterization, and mitigation of threats will be essential to successful submissions.
The Future of Medical Devices and Diagnostics
The future of the medical device and diagnostics industries is tied to smarter and more connected products. To date, these products have demonstrated clear benefits to patients, hospitals, and manufacturers and will continue to improve healthcare distribution and access. Cybersecurity threats posed by medical devices are well documented and will worsen without modernizing device design and quality management processes. As a result, device manufacturers will increasingly be required to mitigate these risks in the products they sell.
To learn more about FDA’s medical device cybersecurity guidance and its impact on your product development plans, please connect with us to start a conversation. Triple Ring has 20 years of experience designing and developing cutting-edge medical technology and a long track record of supporting successful 510(k) clearances and premarket approvals (PMA). We are eager to help you navigate the changes resulting from this FDA guidance.
FAQs
What are the new FDA guidelines for medical device cybersecurity?
FDA’s guidance on Cybersecurity in Medical Devices adds Secure Product Development Frameworks (SPDF) to the risk management processes required of device manufacturers. Risk management is the essential systematic practice of identifying, analyzing, evaluating, controlling, and monitoring risk (now including cybersecurity risk) throughout the product lifecycle. The guidance also describes recommendations regarding the cybersecurity information to be submitted for devices under 510k, PMA, and other submissions.
What does the new FDA guidance on medical device cybersecurity mean for me?
All software-enabled medical devices or in vitro diagnostics featuring connectivity (wireless, local area network, Internet), or portable media (USB or CD) are subject to additional regulatory standards and design controls. If your medical device product fits the above description, you will face additional development effort and development costs to meet the new cybersecurity standards.
How do I design medical devices and IVDs for the new FDA guidance on medical device cybersecurity?
To meet FDA Cybersecurity in Medical Devices guidelines you will need to add Secure Product Development Frameworks (SPDF) like STRIDE to your risk management process. In all of your regulatory submissions, you will also need to address specific elements described in the FDA’s guidance document. Medical Device Engineering consultancies, like Triple Ring Technologies, can help you with these processes.
How does the FDA define connected medical devices?
FDA’s guidance can be interpreted as any device with cybersecurity considerations, including but not limited to devices that have a device software function or that contain software (including firmware) or programmable logic. The guidance also includes devices that are network-enabled or contain other connected capabilities.
Is my medical device product a connected device as defined by the FDA?
The answer is yes if your device contains software, firmware, or programmable logic. Also included are devices that are network-enabled or contain other connected capabilities. Examples of connected devices are MRI systems connected to an internal hospital network, in vitro diagnostics with wireless communications, or implantable devices that can be programmed remotely. Examples of devices that are not connected include orthopedic screws, tongue depressors, and bedpans.



